Differential Expression Analysis for CCC & Downstream Signalling Networks#
Background#
Cell-cell communication (CCC) events play a critical role in diseases, often experiencing deregulation. To identify differential expression of CCC events between conditions, we can build upon standard differential expression analysis (DEA) approaches, such as DESeq2. While dimensionality reduction methods like extracting intercellular programmes with MOFA+ and Tensor-cell2cell reduce CCC into sets of loadings, hypothesis-driven DEA tests focus on individual gene changes, making them easier to understand and interpret.
In this tutorial, we perform DEA at the pseudobulk level to assess differential expression of genes between conditions. We then translate the results into deregulated complex-informed ligand-receptor interactions and analyze their connections to downstream signaling events.
For further information on pseudobulk DEA, please refer to the Differential Gene Expression chapter in the Single-cell Best Practices book, as well as Decoupler’s pseudobulk vignette. These resources provide more comprehensive details on the subject.
Load Packages#
Install mofa, decoupler, and omnipath via pip with the following commands:
pip install "decoupler>=0.1.4"
pip install "pydeseq2>=0.4.0"
import numpy as np
import pandas as pd
import scanpy as sc
import plotnine as p9
import liana as li
import decoupler as dc
import omnipath as op
# Import DESeq2
from pydeseq2.dds import DeseqDataSet
from pydeseq2.ds import DeseqStats
# Obtain TF regulons
net = dc.op.collectri(organism='human', remove_complexes=False, license='academic', verbose=False)
Load & Prep Data#
As a simple example, we will look at ~25k PBMCs from 8 pooled patient lupus samples, each before and after IFN-beta stimulation (Kang et al., 2018; GSE96583). Note that by focusing on PBMCs, for the purpose of this tutorial, we assume that coordinated events occur among them.
This dataset is downloaded from a link on Figshare; preprocessed for pertpy.
adata = li.testing.datasets.kang_2018()
adata
AnnData object with n_obs × n_vars = 24673 × 15706
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'condition', 'cluster', 'cell_type', 'patient', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'sample', 'cell_abbr'
var: 'name'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
Define columns of interest from .obs
Note that we use cell abbreviations because MOFA will use them as labels for the views.
sample_key = 'sample'
groupby = 'cell_abbr'
condition_key = 'condition'
Basic QC#
Note that this data has been largely pre-processed & annotated, we refer the user to the Quality Control and other relevant chapters from the best-practices book for information about pre-processing and annotation steps.
# filter cells and genes
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
Showcase the data#
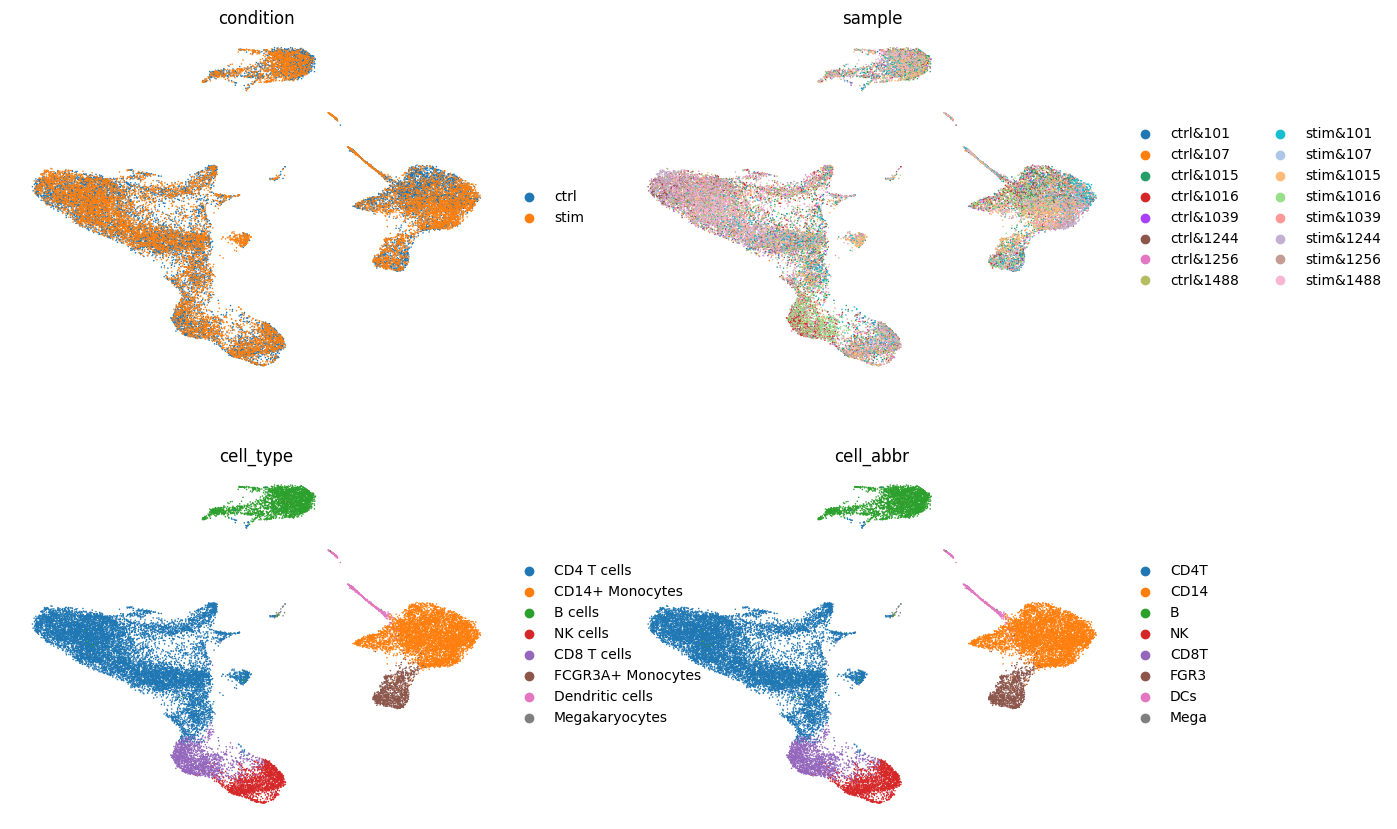
# Show pre-computed UMAP
sc.pl.umap(adata, color=[condition_key, sample_key, 'cell_type', groupby], frameon=False, ncols=2)
Differential Testing#
First, we need to generate pseudobulk profiles for each cell type, and we do so using the decoupler package.
pdata = dc.pp.pseudobulk(
adata,
sample_col=sample_key,
groups_col=groupby,
layer='counts',
mode='sum'
)
pdata
AnnData object with n_obs × n_vars = 110 × 15701
obs: 'condition', 'cell_type', 'patient', 'sample', 'cell_abbr', 'psbulk_n_cells', 'psbulk_counts'
var: 'name', 'n_cells'
layers: 'psbulk_props'
# filter samples based on number of cells and counts
dc.pp.filter_samples(pdata, min_cells = 10, min_counts=1000)
We can plot the quality control metrics for each pseudobulk sample:
dc.pl.filter_samples(pdata, groupby=[sample_key, groupby], figsize=(11, 4))

Differential Expression Analysis#
Next, now that we have generated the pseudobulk profiles, we can perform some edgeR-like filtering using decoupler-py, and then differential expression analysis using the pydeseq2 package - a re-implementation of the original DESeq2 method (Love et al., 2014).
Here, we perform DEA on the pseudobulk profiles for each cell type, for more info check this tutorial: https://decoupler-py.readthedocs.io/en/latest/notebooks/pseudobulk.html
%%capture
dea_results = {}
quiet = True
for cell_group in pdata.obs[groupby].unique():
# Select cell profiles
ctdata = pdata[pdata.obs[groupby] == cell_group].copy()
# Obtain genes that pass the edgeR-like thresholds
# NOTE: QC thresholds might differ between cell types, consider applying them by cell type
genes = dc.pp.filter_by_expr(ctdata,
group=condition_key,
min_count=5, # a minimum number of counts in a number of samples
min_total_count=10 # a minimum total number of reads across samples
)
# Filter by these genes
ctdata = ctdata[:, genes].copy()
# Build DESeq2 object
# NOTE: this data is actually paired, so one could consider fitting the patient label as a confounder
dds = DeseqDataSet(
adata=ctdata,
design_factors=condition_key,
ref_level=[condition_key, 'ctrl'], # set control as reference
refit_cooks=True,
quiet=quiet
)
# Compute LFCs
dds.deseq2()
# Contrast between stim and ctrl
stat_res = DeseqStats(dds, contrast=[condition_key, 'stim', 'ctrl'], quiet=quiet)
stat_res.quiet = quiet
# Compute Wald test
stat_res.summary()
# Shrink LFCs
stat_res.lfc_shrink(coeff='condition_stim_vs_ctrl') # {condition_key}_cond_vs_ref
dea_results[cell_group] = stat_res.results_df
This results in a wall of currently unavoidable verbose text and prints, as such I use %%capture to hide it.
One can use quiet to some of the functions but not logfc_shrinkage
# concat results across cell types
dea_df = pd.concat(dea_results)
dea_df = dea_df.reset_index().rename(columns={'level_0': groupby,'level_1':'index'}).set_index('index')
dea_df.head()
| cell_abbr | baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|---|
| index | |||||||
| AAED1 | B | 7.282872 | -0.134222 | 0.260852 | -0.605923 | 0.544566 | 0.747988 |
| AAMP | B | 11.967019 | -0.436867 | 0.185711 | -2.523263 | 0.011627 | 0.047688 |
| AARS | B | 5.797380 | 0.180074 | 0.272438 | 0.782374 | 0.433995 | 0.660342 |
| AASDHPPT | B | 6.147571 | 0.184129 | 0.305030 | 0.741856 | 0.458175 | 0.679302 |
| AATF | B | 9.567191 | -0.109027 | 0.254673 | -0.493000 | 0.622013 | 0.802867 |
# PyDeseq Seems to intrdoce NAs for some p-values
# NOTE: there sometimes some NaN being introduced, best to double check that, in this case it's only for a single gene, but it might be a problem.
len(dea_df[dea_df.isna().any(axis=1)])
1
DEA to Ligand-Receptor Interactions#
Now that we have DEA results per gene, we can combine them into statistics of potentially deregulated ligand-receptor interactions.
To do so, liana provides a simple function li.multi.df_to_lr that calculates average expression as well as proportions based on the passed adata object, and combines those with the DEA results and a ligand-receptor resource. Since in this case we want to focus on gene statics relevant to the condition (stim), let’s subset the adata to those and normalize the counts.
adata = adata[adata.obs[condition_key]=='stim'].copy()
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
Let’s combine the DEA results with the ligand-receptor interactions. We need to pass the names of the statistics from the DEA table in which we are interest to li.multi.df_to_lr, here we will use the adjusted p-values and Wald test statistic.
lr_res = li.multi.df_to_lr(adata,
dea_df=dea_df,
resource_name='consensus', # NOTE: uses HUMAN gene symbols!
expr_prop=0.1, # calculated for adata as passed - used to filter interactions
groupby=groupby,
stat_keys=['stat', 'pvalue', 'padj'],
use_raw=False,
complex_col='stat', # NOTE: we use the Wald Stat to deal with complexes
verbose=True,
return_all_lrs=False,
)
lr_res = lr_res.sort_values("interaction_stat", ascending=False, key=abs)
lr_res.head()
| ligand | receptor | ligand_complex | receptor_complex | source | ligand_stat | ligand_pvalue | ligand_padj | ligand_expr | ligand_props | ... | receptor_pvalue | receptor_padj | receptor_expr | receptor_props | interaction_stat | interaction_pvalue | interaction_padj | interaction_expr | interaction_props | interaction | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 233 | CXCL11 | CCR5 | CXCL11 | CCR5 | CD14 | 26.172426 | 5.476439e-151 | 2.599815e-148 | 3.106561 | 0.899096 | ... | 9.797079e-47 | 3.410690e-45 | 0.601853 | 0.332369 | 20.264119 | 4.898540e-47 | 1.705345e-45 | 1.854207 | 0.615732 | CXCL11^CCR5 |
| 238 | CCL8 | CCR5 | CCL8 | CCR5 | CD14 | 25.720102 | 6.965891e-146 | 2.798145e-143 | 4.313416 | 0.894756 | ... | 9.797079e-47 | 3.410690e-45 | 0.601853 | 0.332369 | 20.037957 | 4.898540e-47 | 1.705345e-45 | 2.457634 | 0.613562 | CCL8^CCR5 |
| 252 | CCL8 | CCR1 | CCL8 | CCR1 | CD14 | 25.720102 | 6.965891e-146 | 2.798145e-143 | 4.313416 | 0.894756 | ... | 2.567490e-35 | 5.779065e-34 | 1.079367 | 0.552260 | 19.060759 | 1.283745e-35 | 2.889533e-34 | 2.696391 | 0.723508 | CCL8^CCR1 |
| 1062 | CXCL11 | CCR5 | CXCL11 | CCR5 | FGR3 | 16.919574 | 3.227547e-64 | 6.676057e-62 | 2.692778 | 0.826011 | ... | 9.797079e-47 | 3.410690e-45 | 0.601853 | 0.332369 | 15.637693 | 4.898540e-47 | 1.705345e-45 | 1.647316 | 0.579190 | CXCL11^CCR5 |
| 491 | CCL8 | CCR1 | CCL8 | CCR1 | CD14 | 25.720102 | 6.965891e-146 | 2.798145e-143 | 4.313416 | 0.894756 | ... | 2.993682e-07 | 2.388773e-06 | 0.384328 | 0.247232 | 15.421973 | 1.496841e-07 | 1.194386e-06 | 2.348872 | 0.570994 | CCL8^CCR1 |
5 rows × 22 columns
Dealing with heteromeric complexes#
LIANA will filter lowly-expressed interactions, i.e. those for which any of the genes are not expressed in at least 0.1 of the cells (by default) in the AnnData object. This can be adjusted with the expr_prop parameter.
Moreover, to deal with complexes for each cell type, as either source or target of the potential CCC events, LIANA will find and assign the subunit of a complex with the lowest gene expression (by default) as the subunit of interest, and will then use the stats for that subunit as the stats of the whole protein complex.
To this end, we also provide the option to provide a complex_col parameter, which will be used to assign the complex subunit of interest. This column should be a part of the stat_keys. Note that the absolute minimum value is used (i.e. the value closest to 0 is thought to be the ‘worst’ result), so this will not work for statistics with ascending values (e.g. p-values).
Visualize the Results#
interaction_* columns returned by li.multi.dea_to_lr are just the mean of the ligand and receptor columns of the corresponding statistic!
Please use with caution as this is just a summary of the interaction that we can use to e.g. to sort the interactions as done above.
Instead, we recommend to use the ligand and receptor statistics separately to filter and visualize the interactions.
Moreover, by averaging the statistics across the ligand and receptor, we are focusing on the interactions for which both the ligand and receptor are deregulated in the same direction, i.e. both up or both down. However, this might ignore interactions in which e.g. the the ligand is deregulated while the receptor is not, or such where they are deregulated in opposite directions. These could represent potential inhibitory mechanisms, but we leave this to the user to explore.
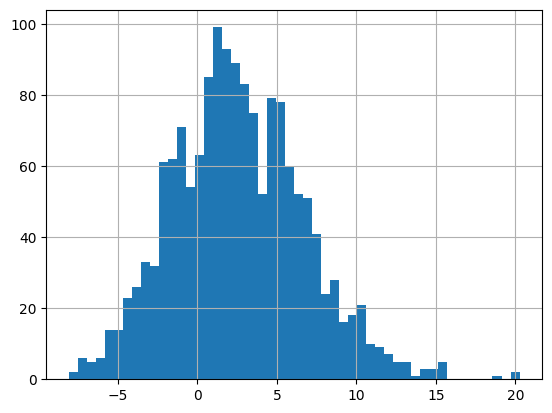
# Let's visualize how this looks like for all interactions (across all cell types)
lr_res = lr_res.sort_values("interaction_stat", ascending=False)
lr_res['interaction_stat'].hist(bins=50)
<Axes: >
Now that we have covered the basics, we can visualize our interactions in a few ways.
Let’s start with the top interactions according to their Wald statistic, and then plot the statistics for the ligands & receptors involved in those interactions across cell types, to do so LIANA+ provide li.pl.tileplot:
li.pl.tileplot(liana_res=lr_res,
fill = 'expr',
label='padj',
label_fun = lambda x: '*' if x < 0.05 else np.nan,
top_n=15,
orderby = 'interaction_stat',
orderby_ascending = False,
orderby_absolute = False,
source_title='Ligand',
target_title='Receptor',
)

<Figure Size: (500 x 500)>
If you want to plot the expression values for ligand-receptor interactions without the DEA statistics, you can set the return_all_lrs parameter to True in the li.multi.dea_to_lr function. This will return a dataframe with all the ligand-receptor interactions, where missing DEA stats will be set as nan, while mean expression and proportions per cluster will be obtained via the AnnData object.
Ligand-Receptor Plot#
We can also use visualize of the stats, summarized at the level of the interaction, to prioritize the interactions, or any subunit statistics using li.pl.dotplot. For example, we can visualize the mean Wald statistic between the ligand & receptor, together with the pvalues for the ligand.
plot = li.pl.dotplot(liana_res=lr_res,
colour='interaction_stat',
size='ligand_pvalue',
inverse_size=True,
orderby='interaction_stat',
orderby_ascending=False,
orderby_absolute=True,
top_n=10,
size_range=(0.5, 4)
)
# customize plot
(
plot
+ p9.theme_bw(base_size=14)
# fill cmap blue to red, with 0 the middle
+ p9.scale_color_cmap('RdBu_r', midpoint=0, limits=(-10, 10))
# rotate x
+ p9.theme(axis_text_x=p9.element_text(angle=90), figure_size=(11, 6))
)

<Figure Size: (1100 x 600)>
Now that we have identified a set of interactions that are potentially deregulated we can look into the downstream signalling events that they might be involved in.
Intracellular Signaling Networks#
Cellular signaling networks govern the behavior of cells, allowing them to respond to external signals, including various cell-cell communication events. Thus, CCC events can be thought of as upstream perturbants of intracellular signaling networks that lead to deregulations of downstream signaling events. Such deregulations are expected to be associated with various conditions and disease. Thus, understanding intracellular signaling networks is critical to model the cellular mechanisms.
Here, we will combine several tools to identify plausible signalling cascades driven by CCC events.
Our approach includes the following steps:
Select a number of potentially deregulated ligand-receptor interactions (input nodes), in terms of summarized PyDESeq2 statistics.
Select a number of potentially deregulated TFs (output nodes). This is done via the use of Transcription factor (TF) activity inference. Carried out on differential gene expression data using TF regulon knowledge with decoupler
Obtain a prior knowledge network (PKNs), with signed protein-protein interactions from OmniPath.
Generate weights for the nodes in the PKN
Use CORNETO to identify a solution in the form of a causal (smallest sign-consistent signaling) network that explains the measured inputs and outputs

Import OmniPath#
For this part OmniPath is required.
# utily function to select top n interactions
def select_top_n(d, n=None):
d = dict(sorted(d.items(), key=lambda item: abs(item[1]), reverse=True))
return {k: v for i, (k, v) in enumerate(d.items()) if i < n}
Select Cell types of Interest#
One limitation of using DEA to identify interactions of interest is that it tells us little about deregulation at the level of cell types. However, from dimensionality reductions on CCC, as done with Tensor-cell2cell & MOFA on the same dataset, we can see there is a potential deregulation of CCC that involve CD14 monocytes both as sources (senders) and targets (or receivers) of intecellular communication. Thus, we will focus on the interactions and downstream signalling within that cell type.
source_label = 'CD14'
target_label = 'CD14'
# NOTE: We sort by the absolute value of the interaction stat
lr_stats = lr_res[lr_res['source'].isin([source_label]) & lr_res['target'].isin([target_label])].copy()
lr_stats = lr_stats.sort_values('interaction_stat', ascending=False, key=abs)
Select Receptors based on interaction stats#
These will be used as the input or start nodes for the network. In this case, we will use interactions potentially involved in autocrine signalling in CD14 monocytes.
lr_dict = lr_stats.set_index('receptor')['interaction_stat'].to_dict()
input_scores = select_top_n(lr_dict, n=10)
input_scores
{'CD40': 15.213890738775456,
'CD80': 11.672283052638337,
'SIRPA': 9.92679527367514,
'CCR1': 9.12729237916692,
'HLA-DPB1': 8.625121687728944,
'LILRB2': 8.250712282572682,
'CCR5': 6.024106443619973,
'PTPRC': 5.811339038637888,
'LILRB1': 5.586746654720494,
'CD47': 5.431274692175629}
Select Transcription Factors of interest#
Before we select the transcription factors, we need to infer their activity. We will do so using decoupler with CollecTri regulons. Specifically, we will estimate TF activities using the Wald statistics (from PyDESeq2) for the genes in the regulons.
# First, let's transform the DEA statistics into a DF
# we will use these to estimate deregulated TF activity
dea_wide = dea_df[[groupby, 'stat']].reset_index(names='genes').pivot(index=groupby, columns='genes', values='stat')
dea_wide = dea_wide.fillna(0)
dea_wide
| genes | A1BG | AAAS | AAED1 | AAGAB | AAK1 | AAMDC | AAMP | AAR2 | AARS | AARSD1 | ... | ZSCAN16 | ZSCAN16-AS1 | ZSCAN18 | ZSCAN32 | ZSWIM6 | ZSWIM7 | ZUFSP | ZW10 | ZYX | ZZZ3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_abbr | |||||||||||||||||||||
| B | 0.000000 | 0.000000 | -0.605923 | 0.000000 | 0.000000 | 0.000000 | -2.523263 | 0.000000 | 0.782374 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | -2.350376 | 0.000000 | 0.00000 | 0.000000 | 0.000000 |
| CD14 | -2.993111 | 0.000000 | 10.129553 | 0.000000 | 1.085436 | 6.968489 | -3.097932 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | -1.281381 | -4.154833 | 1.737279 | 0.00000 | -1.796965 | 0.000000 |
| CD4T | -2.814957 | -1.995631 | 0.191978 | 1.144712 | -5.150155 | -0.081141 | -0.650599 | 2.032146 | 1.648662 | -0.270347 | ... | -0.218578 | -1.168355 | -1.515016 | -0.430795 | 0.000000 | -1.685312 | 3.455590 | -0.17713 | -1.399221 | -0.240491 |
| CD8T | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 | 0.000000 | 0.000000 |
| DCs | 0.000000 | 0.000000 | 3.883796 | 0.000000 | 0.000000 | 0.000000 | -1.620856 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 | -2.573142 | 0.000000 |
| FGR3 | 0.000000 | 0.000000 | 2.836840 | 0.000000 | 0.000000 | 0.000000 | -3.248816 | 0.000000 | 0.000000 | 0.000000 | ... | 4.160610 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 | -2.117717 | 0.000000 |
| NK | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | -2.548184 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 | 0.000000 | 0.000000 |
7 rows × 7802 columns
# Run Enrichment Analysis
estimates, pvals = dc.mt.ulm(mat=dea_wide, net=net)
estimates.T.sort_values(target_label, key=abs, ascending=False).head()
| B | CD14 | CD4T | CD8T | DCs | FGR3 | NK | |
|---|---|---|---|---|---|---|---|
| STAT1 | 17.926670 | 13.178566 | 16.732069 | 17.533529 | 17.887823 | 15.023597 | 16.353252 |
| IRF1 | 15.773025 | 10.694732 | 14.545148 | 13.118623 | 14.475640 | 11.968679 | 13.322178 |
| STAT2 | 14.908431 | 9.841800 | 12.172835 | 13.719251 | 15.724816 | 12.524464 | 12.979574 |
| IRF9 | 18.450977 | 8.841106 | 14.894291 | 19.616129 | 17.128387 | 13.703831 | 17.811499 |
| NFKB1 | 6.898114 | 7.734655 | 8.250937 | 5.890799 | 9.258309 | 8.580269 | 7.130418 |
Select top TFs#
Now that we have the potentially deregulated TFs, we focus on the top 10 TFs, based on their enrichment scores. In this case, we will look specifically at the top TFs deregulated in CD14 monocytes.
tf_data = estimates.copy()
tf_dict = tf_data.loc[target_label].to_dict()
output_scores = select_top_n(tf_dict, n=5)
Generate a Prior Knowledge Network#
Now we will obtain protein-protein interactions from OmniPath, filter them according to curation effort to ensure we only keep those that are of high quality, and convert them into a knowledge graph.
# obtain ppi network
ppis = op.interactions.OmniPath().get(genesymbols = True)
ppis['mor'] = ppis['is_stimulation'].astype(int) - ppis['is_inhibition'].astype(int)
ppis = ppis[(ppis['mor'] != 0) & (ppis['curation_effort'] >= 5) & ppis['consensus_direction']]
input_pkn = ppis[['source_genesymbol', 'mor', 'target_genesymbol']]
input_pkn.columns = ['source', 'mor', 'target']
input_pkn.head()
| source | mor | target | |
|---|---|---|---|
| 3 | CAV1 | 1 | TRPC1 |
| 6 | ITPR2 | 1 | TRPC1 |
| 9 | STIM1 | 1 | TRPC1 |
| 10 | TRPC1 | 1 | TRPC3 |
| 11 | TRPC3 | 1 | TRPC1 |
# convert the PPI network into a knowledge graph
prior_graph = li.mt.build_prior_network(input_pkn, input_scores, output_scores, verbose=True)
In this section we use Prior Knowledge Networks (PKNs) from OmniPath to generate network hypotheses based on the deregulated interactions considering both sign and direction. Specifically, we focus on highly curated protein-protein interactions, which often represent hubs in the network. Since such network approaches are highly dependent on prior knowledge, for a review on prior knowledge bias and similar network inference methods, including thier limitations, see Garrido-Rodriguez et al., 2022.
Calculate Node weights#
Calculate gene expression proportions within the target cell type; we will use those as node weights in the network.
temp = adata[adata.obs[groupby] == target_label].copy()
node_weights = pd.DataFrame(temp.X.getnnz(axis=0) / temp.n_obs, index=temp.var_names)
node_weights = node_weights.rename(columns={0: 'props'})
node_weights = node_weights['props'].to_dict()
Find Causal Network#
CORNETO (Rodriguez-Mier et al., In prep) generalizes biological network inference problems using convex and combinatorial optimization. Here, we use it to find the smallest sign-consistent network that explains the measured inputs and outputs, a network inference problem formulated in CARNIVAL.
To run CORNETO, we need to first install it; it’s very lightweight and can be installed via pip:
pip install corneto==0.9.1-alpha.6 cvxpy cylp
import corneto as cn
cn.info()
 |
|
df_res, problem = li.mt.find_causalnet(
prior_graph,
input_scores,
output_scores,
node_weights,
# penalize (max_penalty) nodes with counts in less than 0.1 of the cells
node_cutoff=0.1,
max_penalty=1,
# the penaly of those in > 0.1 prop of cells set to:
min_penalty=0.01,
edge_penalty=0.1,
verbose=False,
max_runs=50, # NOTE that this repeats the solving either until the max runs are reached
stable_runs=10, # or until X number of consequitive stable runs are reached (i.e. no new edges are added)
solver='gurobi' # 'scipy' is available by default, but often results in suboptimal solutions
)
Set parameter Username
Academic license - for non-commercial use only - expires 2025-01-26
Visualize the Inferred Network#
Now that the solution has been found, we can visualize it using the cn.methods.carnival.visualize_network function.
cn.methods.carnival.visualize_network(df_res)

We can see that the network above, largely captures a potential regulatory cascade with inhibitory (–|) and stimulatory (–>) interactions, related to JAK-STAT signalling. The network, in this case, starts from a receptor (triangle), coming from the top interactions, and ends with the deregulated TFs (square). The remainder of the nodes (circles) were inferred, taking their weights into account, and were not necessarily included in the input or output nodes.
In this example, we represent the directionality of signalling such that intracellular signalling is downstream of intercellular communication events. However, in biology cellular response is an admixture of both; thus such approaches are a simplification of biological reality.
Describe Results#
Let’s examine the result of the subnetwork search - it provides information about the predicted signs of nodes and edges.
df_res.head()
| source | source_type | source_weight | source_pred_val | target | target_type | target_weight | target_pred_val | edge_type | edge_pred_val | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PRKCZ | unmeasured | 0.0 | 1.0 | JAK1 | unmeasured | 0.000000 | 1.0 | 1 | 1.0 |
| 1 | STAT1 | unmeasured | 0.0 | 1.0 | IRF1 | output | 10.694732 | 1.0 | 1 | 1.0 |
| 2 | PRKACA | unmeasured | 0.0 | 1.0 | NFKB1 | output | 7.734655 | 1.0 | 1 | 1.0 |
| 3 | SRC | unmeasured | 0.0 | 1.0 | PDPK1 | unmeasured | 0.000000 | 1.0 | 1 | 1.0 |
| 4 | GNAI2 | unmeasured | 0.0 | 1.0 | SRC | unmeasured | 0.000000 | 1.0 | 1 | 1.0 |
Nodes#
source: source nodes in the Protein-Protein Interaction (PPI) network. Suffixes such as “_s,” “_pert_c0,” and “_meas_c0” indicate specific experimental conditions or measurement types (they are there simply because of how the ILP problem is formulated, and can be ignored).
target: target nodes in the PPI network, with suffixes similar to the source node.
source_type (unmeasured, input):
input: start nodes (provided by the users, here receptors).
output: end nodes (provides by the user, here transcription factors).
unmeasured: Nodes that are neither input nor output - i.e. those that predicted by the algorithm.
source_weight and target_weight: Inputs to the causal net method, indicating the influence of “measured” nodes within the network. Only the sign is taken into account.
source_pred_val (1, 0, -1): Regulatory state of the node:
1: Upregulated
0: No differential expression
-1: Downregulated
target_pred_val (1, -1): Regulatory state of the target node:
1: Upregulated
-1: Downregulated
Edges (interaction)#
edge_type (1, -1, 0): Type of interaction from prior knowledge:
1: Activating interaction (e.g., A -> B)
-1: Inhibitory interaction
edge_pred_val (1, -1): Predicted effect of the interaction on the target node:
1: Upregulation
-1: Downregulation
Installing the Gurobi Solver: A Step-by-Step Guide#
While in this small example the internal scipy solver works, for larger networks we recommend using a solver such as Gurobi.
Gurobi is a powerful optimization solver used in various mathematical programming problems. Here’s how you can install it:
1. Download Gurobi for Your Operating System:#
Visit the Gurobi download page and select the version compatible with your OS.
2. Unzip and Update Path:#
After downloading, unzip the file. Locate the /bin folder inside the unzipped directory and add it to your system’s $PATH variable. This step is crucial as it allows your system to recognize and run Gurobi from anywhere.
3. Register for an Academic License:#
If you’re an academic user, you can obtain a free license. Register and request an academic license through the Gurobi portal. Follow the prompts to complete your registration.
4. Install Gurobi Python Interface:#
Open your command prompt or terminal and run:
pip install gurobipy
This command installs the necessary Python interface to interact with Gurobi.
5. Configure the Solver:#
In your code, ensure the solver parameter is set to gurobi to direct your program to use the Gurobi solver.
By following these steps, you should have Gurobi installed and ready to tackle complex optimization problems.